

近日,公司何建平教授团队与华东师范大学汤静教授、夏伟博士以及日本早稻田大学Yusuke Yamauchi教授等人合作在化学领域的权威期刊《J. Am. Chem. Soc.》发表有关氧还原催化剂的高水平论文。第一作者为开云Kaiyun博士研究生李晶晶。具体研究成果参见“Metal–Organic Framework-Derived Graphene Mesh: a Robust Scaffold for Highly Exposed Fe–N4 Active Sites toward an Excellent Oxygen Reduction Catalyst in Acid Media”。
论文链接:https://pubs.acs.org/doi/full/10.1021/jacs.2c00719
全文速览
通过熔盐剥离(氯化锂和氯化钾分别作为蚀刻剂和剥离剂)策略, 成功将片状的类沸石咪唑骨架(Zn-ZIF-L)热剥离为超薄的氮掺杂的类石墨烯筛 (NGM)。 通过调节热剥离条件,可以精细地调节NGM的孔隙大小,同时分散和锚定Fe-N4位点,以获得具有高度暴露且高负载的铁单原子催化剂(SA-Fe-NGM)。独特的二维形态、高孔隙度、缺陷石墨烯周围丰富且可接近的Fe-N4活性位点等特点,使得SA-Fe-NGM催化剂在酸性电解液中的氧还原半波电位达到0.83 V。本研究突破传统三维金属有机框架(MOF)衍生碳的尺寸限制,制备新型低维 MOF 衍生功能碳材料,并提出实现高金属负载和高暴露的单原子催化剂的精细策略。
背景介绍
Fe-N-C催化剂在酸性条件下的氧还原(ORR)催化性能优异,特别是含有Fe-Nx结构的单原子催化剂(Fe-SAC)能够进一步提高ORR催化性能。但是相比于贵金属铂基催化剂,Fe-SAC催化剂在半波电位和电流密度方面仍存在明显差距。Fe-SAC催化剂的性能与碳载体的结构密切相关。目前,金属有机框架(MOFs)被广泛用作Fe-SAC催化剂的理想前驱体,由MOF衍生的催化剂大多为三维结构,相比于埋藏在致密碳基体中的Fe-Nx位点, 靠近催化剂外表面的由微孔锚定的Fe-Nx位点参与ORR反应的机会更多。理想情况下,只有导电碳基三相界面(TPI)上的Fe-Nx位点才能作为真正的活性位点参与ORR反应。二维多级孔碳材料相较于三维碳材料能够最大限度地锚定Fe-Nx位点,且介孔结构提供快速的传质通道,显著增加TPI数量。因此,构建具有高密度活性位点、超暴露纳米结构和分级多孔结构的Fe-SAC催化剂是提高活性位点利用率和ORR催化性能的有效策略。
本文亮点
1. 实现超薄二维氮掺杂类石墨烯筛材料(NGM)的孔径分布可调整目标。采用熔盐剥离策略,改变氯化锂与氯化钾的投料比,在惰性氛围下高温热解片状Zn-ZIF-L获得不同孔径分布的NGM。为后续金属大环化合物的负载提供良好的碳载体环境;
2. 实现了高金属且高密度Fe-Nx位点的单原子Fe催化剂(SA-Fe-NGM)的构建。碳载体负载的金属含量高达8.36 wt%, 在0.5M的H2SO4电解液中,SA-Fe-NGM催化剂表现出优异的催化性能,半波电位达到0.83 V;
3. 结合第一性原理计算和碳基底的孔径分布分析,位于孔缺陷处的Fe-N4活性位点更有利于催化ORR过程。
图文解析
图1为NGM, FePc-NGM和SA-Fe-NGM材料的合成过程。首先,具有层状结构的Zn-ZIF-L为剥离成超薄二维材料提供可能性,在KCl和LiCl混合盐与Zn-ZIF-L前驱体混合后,经过900 ℃氩气氛围下生成NGM。在这个过程中,K+离子能够嵌入Zn-ZIF-L材料中实现剥离,Li+离子有助于腐蚀由Zn-ZIF-L衍生的碳材料,形成介孔和大孔结构。随后,将金属大环化合物FePc分子负载在NGM上,获得FePC-NGM复合材料,然后对其进行惰性氛围下的高温热解获得具有高负载和高暴露的FeN4活性位点的单原子催化剂SA-Fe-NGM。
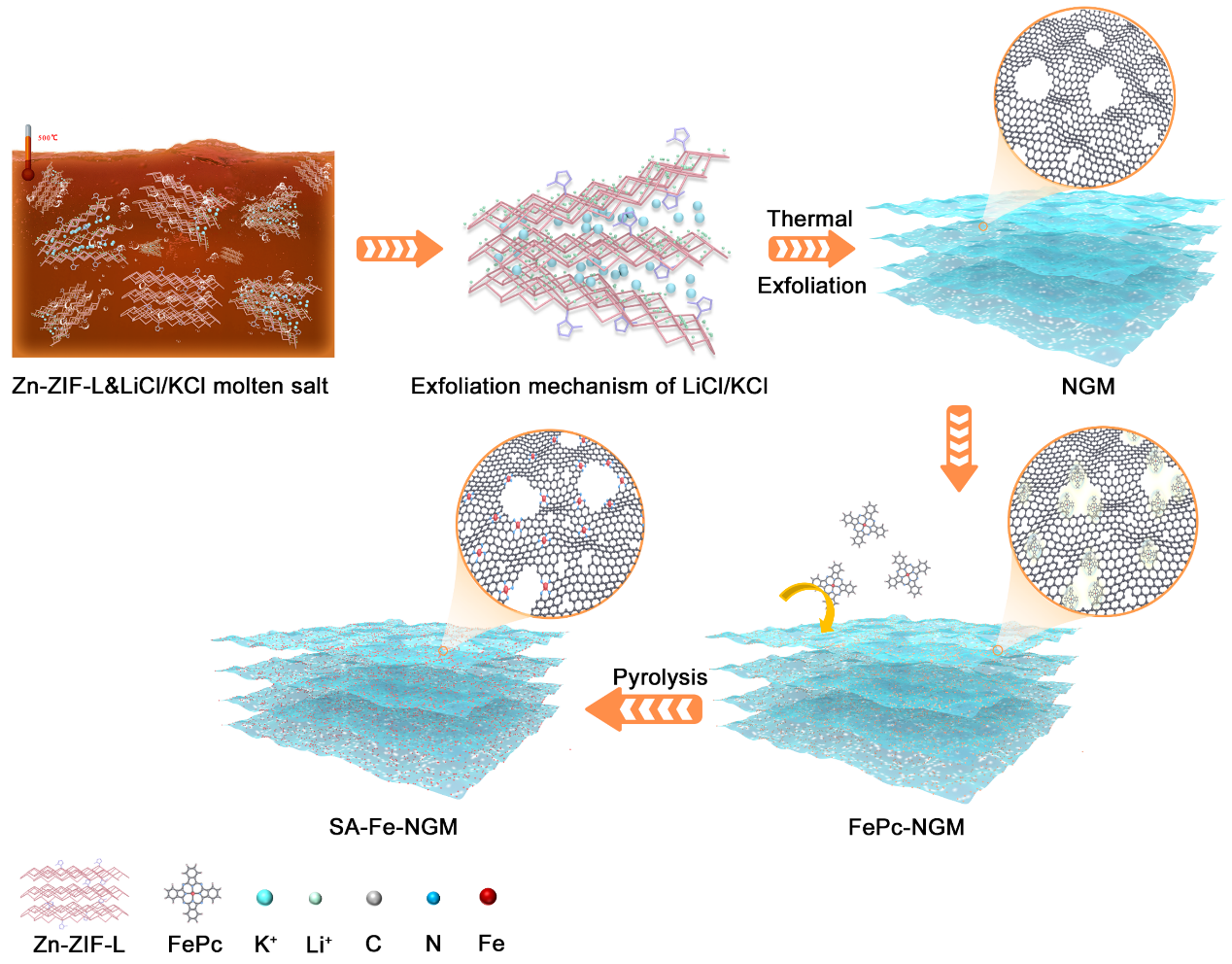
图1 SA-Fe-NGM催化剂的合成示意图
相比于完美石墨烯,碳纳米管和炭黑等,NGM作为碳载体负载金属具有巨大优势。在该材料的合成过程中,通过调整LiCl和KCl的投料比获得不同比表面积和孔径分布的NGM。如图2(a-c)所示,热剥离过程中LiCl/KCl质量比的变化导致NGM孔隙度发生明显变化,表现出从微孔到中孔的不同等级的孔隙结构。其中负载了FePc后的NGM-Li+/K+ = 0.2的孔径分布模式与NGM-Li+/K+ =0.2不同,孔径位置发生偏移。如图2b所示,FePc-NGM-Li+/K+ = 0.2的孔体积数量的增加集中在1.5 ~ 3.4 nm范围,可能是FePc分子吸附在孔径范围为2.5 ~ 5.0 nm的介孔附近,导致该范围的孔径尺寸减小。后续热解过程中,SA-Fe-NGM-Li+/K+ = 0.2在~ 3.4 nm孔径处的孔体积与NGM-Li+/K+ = 0.2相近,是由于负载在该孔径处不稳定的FePc分子经过高温热处理全部分解。而孔径范围小于2.0 nm的微孔体积变化较小,表明Fe-Nx位点在该孔径范围内可能得到了稳定构建。当LiCl与KCl的质量比为0.4时,NGM-Li+/K+ = 0.4的平均孔径为5.04 nm, 较多的FePc分子吸附在该孔径周围导致FePc分子堆积,经高温热解后可能形成较大的Fe纳米团簇导致部分介孔被堵塞(图2c)。当LiCl与KCl的质量比为0.1时,FePc分子在NGM-Li+/K+ = 0.1表面的吸附主要是共轭吸附而不是锚定在多孔周围,这种作用导致高温热解过程中复合物不稳定,易分解(图2a)。通过高分辨率的高角度环形暗场扫描透射电镜观察SA-Fe- NGM-Li+/K+ = 0.1和SA-Fe- NGM-Li+/K+ = 0.2中的Fe以单原子形式分散在碳基底中,其中SA-Fe- NGM-Li+/K+ = 0.2的Fe单原子密度更高,而SA-Fe- NGM-Li+/K+ = 0.4中存在Fe纳米团簇(图2d-f),这个结果进一步表明不同孔径分布的NGM对Fe金属负载的影响。此外,NGM具有较大的二维平面和丰富的多级孔结构,可以推断Fe-N4位点可能被锚定在碳基底的平面,孔空位或边缘处。如图2(g-i)所示,位于孔空位处的Fe-N4位点的形成能最低,表明孔空位在热力学上更有利于Fe-N4位点的稳定。
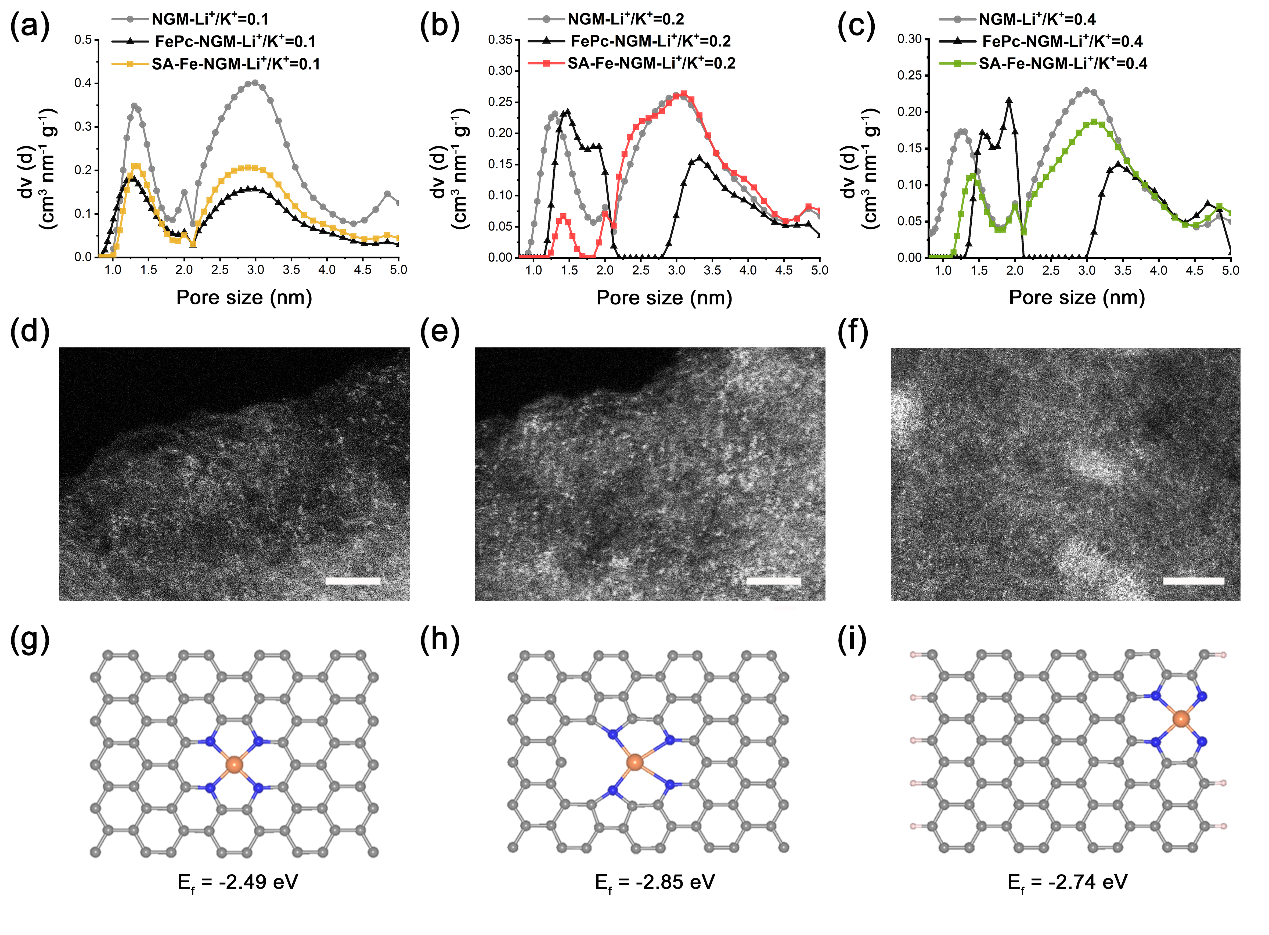
图2 不同LiCl/KCl比例下的催化剂 (a-c)孔径分布图,(d-e)HAADF-STEM图。SA-Fe-NGM催化剂中Fe-N4位点锚定的三个位置(g)平面,(h)空位,(i)边缘的形成能。
选取孔径分布最优的SA-Fe-NGM-Li+/K+ = 0.2(简写为SA-Fe-NGM)为对象,对其形貌结构和物相结构进行深入研究。如图3c所示, 在透射电镜下观察到SA-Fe-NGM呈现出平面尺寸为6 μm ⅹ 9 μm的超薄二维结构。在高倍电镜下观察到该材料中存在随机取向的石墨化碳和高缺陷碳结构。如图3e所示,利用原子力显微镜表征SA-Fe-NGM的厚度大约为1.5 nm。

图3 SEM图(a)Zn-ZIF-L, (b)SA-Fe-NGM;SA-Fe-NGM的(c)低倍TEM图,(d)高倍TEM图,(e)HAADF-STEM图,(f)原子力显微镜图。
利用XAS分析SA-Fe-NGM的局部配位结构。如图4a所示,Fe k边XANES揭示了样品中Fe的价态为+2 ~ +3. EXAFS分析样品在1.47 Å位置存在的信号峰属于Fe-N(O),表明Fe周围的配位原子可能是N原子或O原子(图4b)。进一步对EXAFS进行傅里叶变换拟合分析,结果表明与Fe-N(O)对应的平均配位数约为5(图4c)。 为了确定活性位点的配位结构,我们对Fe k边XANES的实验光谱和理论光谱进行了计算(图4e),结果表明SA-Fe-NGM的活性位点结构为平面Fe-N4结构,在轴向端式吸附一个O2分子。
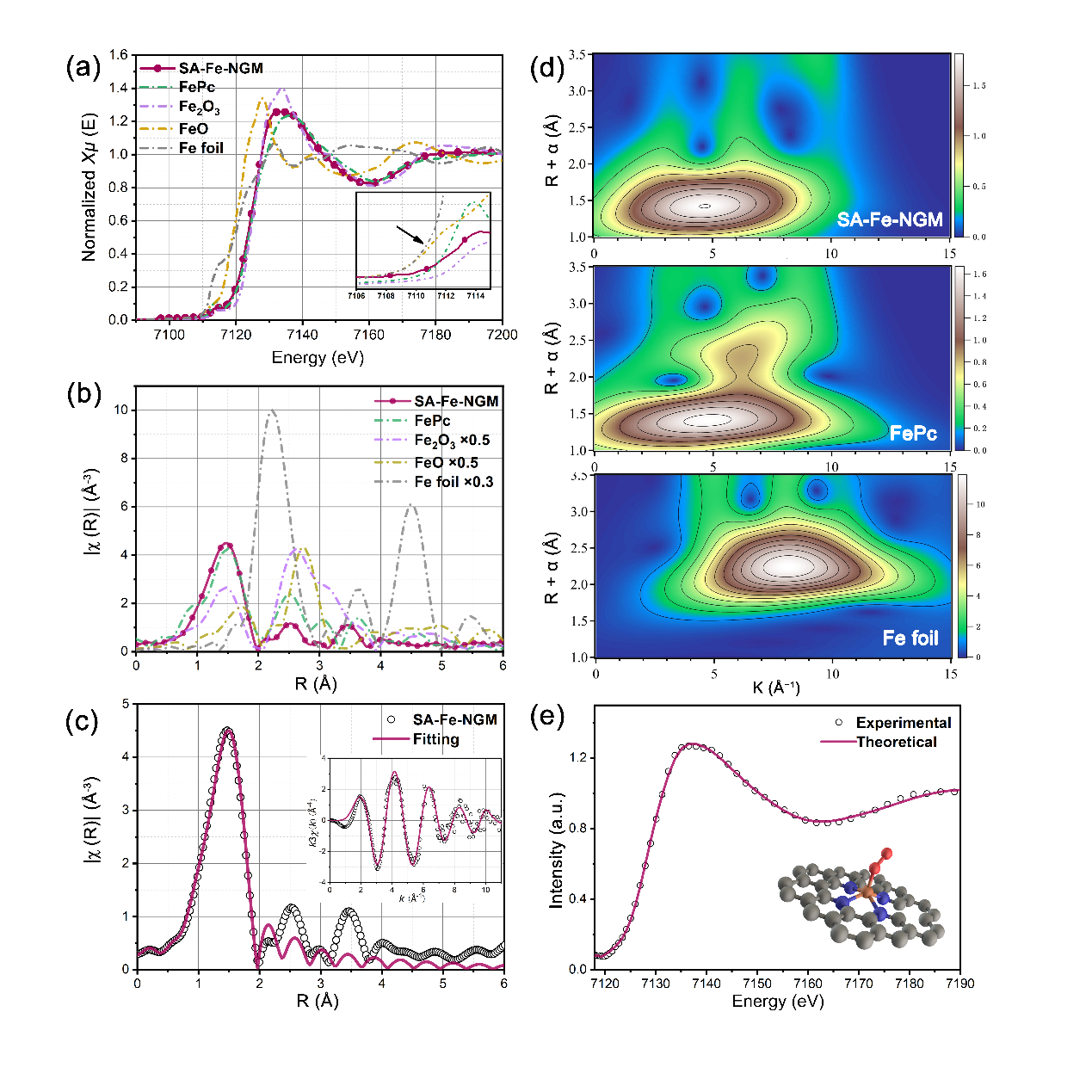
图4. SA-Fe-NGM催化剂的 (a)Fe-k边XANEs谱,(b)EXAFs谱,(c)傅里叶转换EXAFs拟合,(d)小波转换图,(e)Fe k边XANEs的实验谱与理论谱计算。
在0.5 M H2SO4电解液中对SA-Fe-NGM等样品的催化性能进行评估。结合LSV曲线和塔菲尔斜率分析,SA-Fe-NGM催化剂在酸性条件下的半波电位达到0.83 V vs RHE, 塔菲尔斜率为76.24 mV dec-1(图5a-b)。与一些报道的非贵金属掺杂的碳基电催化剂相比,SA-Fe-NGM催化剂表现出竞争性的ORR活性(图5f)。在0.10.8 V的电位下,SA-Fe-NGM催化剂的H2O2产率小于3.6%,转移电子数高达3.95,表明该催化剂具有良好的选择性和直接4电子过程(图5c)。对催化剂进行加速耐久性测试,在0.5M H2SO4电解液中循环5000圈后,SA-Fe-NGM催化剂的半波电位仅负移了20 mV,表明该催化剂具有良好的稳定性。以该催化剂为阴极进行了H2-O2燃料电池的膜电极组装测试。如图5e所示,膜电极的功率密度最高达到634 mW cm-2。
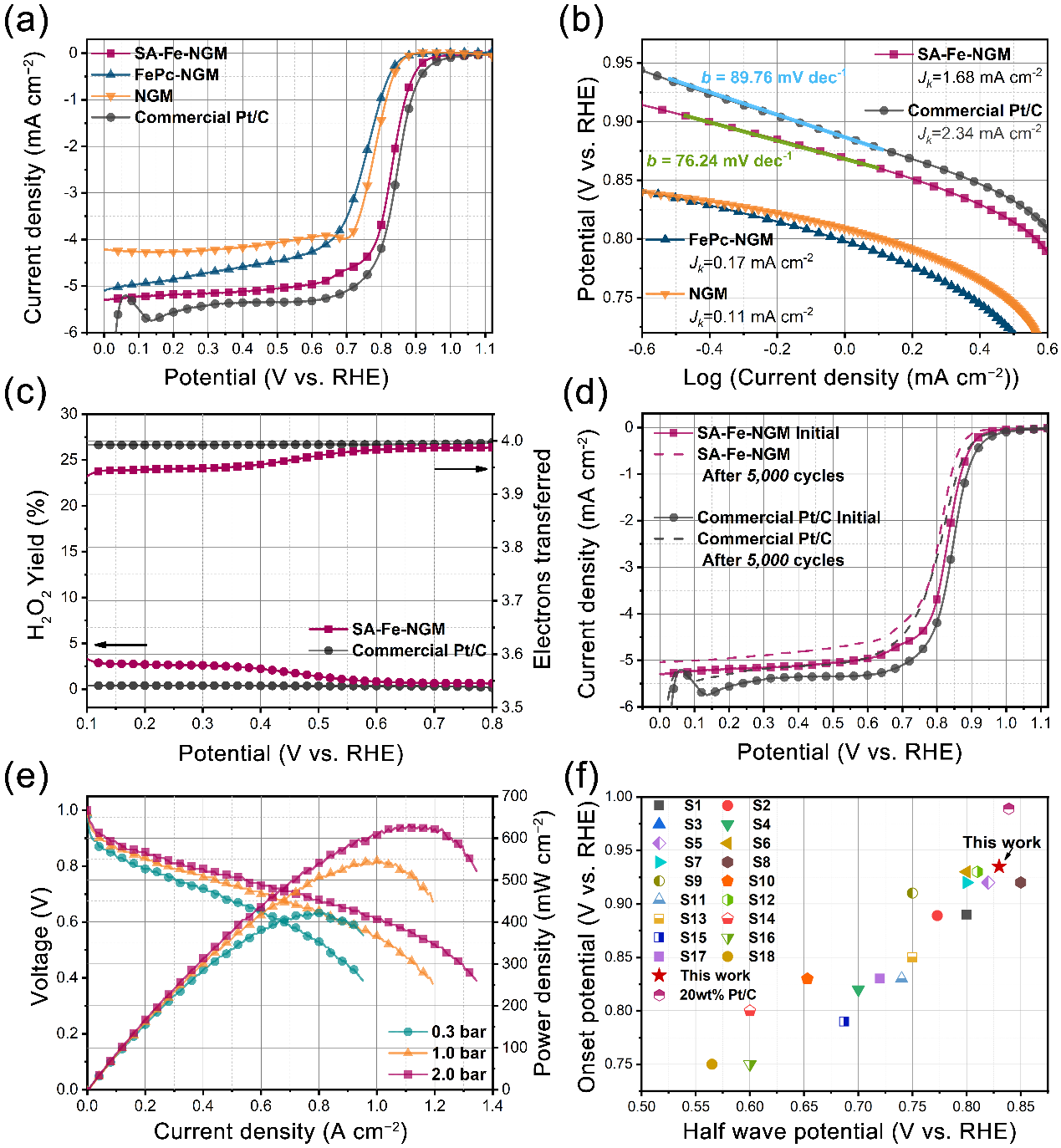
图5. 不同催化剂的(a)LSV曲线,(b)Tafel斜率,(c)H2O2产率和转移电子数,(d)长期循环稳定性,(e)H2-O2膜电极的功率密度图,(f)文献性能对比图。
利用DFT计算进一步研究SA-Fe-NGM催化剂中最优活性位点的ORR催化机制。考虑到SA-NGM-Fe催化剂中存在大量的孔结构和较大的平面二维结构,我们分析锚定Fe-N4活性位点的位置可能有平面,孔空位和边缘。通过ORR自由能变化路径计算表明孔空位锚定的Fe-N4位点的过电位最低(图6a-b)。且空位处的Fe-N4位点的电荷再分布很明显(图6c),表明空位处的电子给与使Fe中心的正电荷降低。研究Fe 3d的态密度,相较于平面处和边缘处的FeN4位点,空位处的FeN4位点中Fe 3d的d带中心位置最负,表明ORR反应的中间体OH*在Fe-N4 (空位)处的吸附能变弱。这些结果表明,锚定在空位处的Fe-N4位点比其他两个位置处的活性更高,能够有效降低反应能垒,对SA-Fe-NGM催化剂的性能提高起着至关重要的作用。
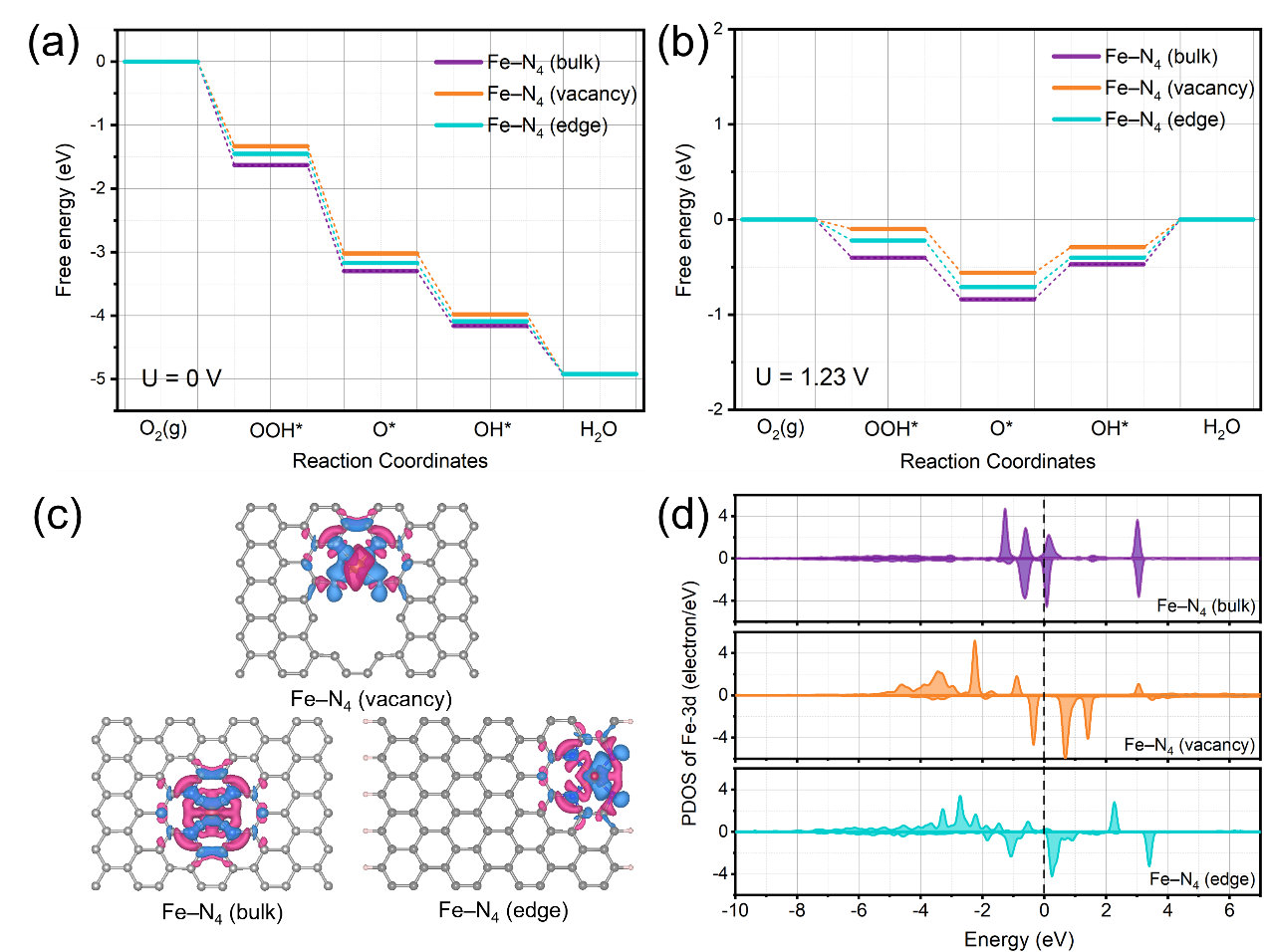
图6. Fe-N4(bulk), Fe-N4(vacancy)和Fe-N4(edge)的ORR自由能变化路径图(a)U = 0V, (b) U=1.23V; (c)差分电荷图,(d)Fe 3d的态密度分布图。

